
#NephJC Chat
Tuesday June 5 9 pm Eastern
Wednesday June 6 8 pm BST, 12 noon Pacific
Am J Hum Genet. 2018 May 3;102(5):816-831. doi: 10.1016/j.ajhg.2018.03.014. Epub 2018 Apr 26.
Patient-iPSC-Derived Kidney Organoids Show Functional Validation of a Ciliopathic Renal Phenotype and Reveal Underlying Pathogenetic Mechanisms.
Forbes TA, Howden SE, Lawlor K, Phipson B, Maksimovic J, Hale L, Wilson S, Quinlan C, Ho G, Holman K, Bennetts B, Crawford J, Trnka P, Oshlack A, Patel C, Mallett A, Simons C, Little MH.
PMID: 29706353 Full Text at AHJG (till June 6th)




Introduction
A large majority of children and at least 10% of adults who receive renal replacement therapy have an inherited kidney disease. The exact genetic basis of a majority of these cases of end stage kidney disease (ESKD) remains obscure. In patients where the exact genetic mutation is unknown much of what we know about the pathogenesis comes from animal models. One potential barrier to the successful use of these models is the significant differences between animal and human genomes. This limits the ability to transform animal models discoveries into patient therapies. To overcome this, Forbes et al in Australia have shown for the first time that kidney disease can be modelled in a dish. But before we get to the guts of this paper there are a few basic concepts to be grasped.
What is an induced pluripotent stem cell (iPSC)?
In 2006, Shinya Yamanaka of Kyoto University, Japan received the Nobel Prize for work that caused' adult mouse cells to ‘forget’ their differentiation and be reprogrammed to an embryonic-like state from where they can grow into any type of cell (pluripotency) by altering just 4 genes. In 2007, this was extended to human adult cells leading to the term human induced pluripotent stem cells (iPSC). These cells, once given the correct environment (growth factor signals), can recapitulate nephrogenesis and are able to self-organize into organoids.
What is an organoid?
An organoid is a 3-dimensional miniature version of an organ which exhibits the following properties:
Has multiple organ-specific cell types (realistic microanatomy),
Is capable of performing some of the organ’s specific functions (e.g. filtration, secretion),
Cells are grouped and spatially organized in a pattern similar to the organ.
Organoids provide an opportunity to recreate the disease in the petri dish which can be studied to better understand the pathophysiology and identify possible treatments.
How iPSCs can be used for disease modeling?
Stem cells (iPSCs) can be tailored using CRISPR/Cas9. It is a gene-editing platform derived from Streptococcus pyogenes using an endonuclease (Cas9) and a synthetic guide RNA to introduce a double-strand DNA segmet at a specific location within the genome. It was a #NephMadness 2017 finalist, read more here. Two types of cells can be used -
When the pathogenic mutations are known (ADPKD, FSGS), these targeted mutations can be induced in human iPSCs. These CRISPR-modified pluripotent stem cells will subsequently grow into human kidney organoids and serve as disease specific models.
But more often, the causative gene is not known. We may have some clues from a family history that can be further elucidated using next generation sequencing or related techniques. Once a novel mutation is identified, it is often unclear how the gene relates to the phenotype. These variants of unknown significance’ (VUS) can be investigated by creating kidney organoids with the VUS using iPSCs derived from the patient. This means literally creating a disease model for an individual patient.
The Study
Methods and Results
This study illustrates for the first time the journey of proband derived iPSCs to kidney organoids bearing the disease phenotype. They also demonstrate an important feature, rectification of the phenotype in the gene corrected organoid, thereby verifying the genomic variant as disease causing.
The patient was diagnosed with retinitis pigmentosa at 1 year of age. At 6 yrs of age, she progressed to ESKD. Phenotypically, this resembled Senior Løken syndrome (SLS). There was no evidence of renal cysts. Her parents were non-consanguineous and there was no family history of kidney disease.
Trio (of proband and both parents) exome sequencing analysis identified a compound heterozygous mutation in single-base-pair of IFT140 gene. This gene codes for a protein involved in the formation and maintenance of cilia. There were two mutations which existed in trans:
- c.634G>A (p.Gly212Arg) and
- c.2176C>G (p.Pro726Ala).
[c.634G>A means that in coding DNA at 634 position from 5’ end guanine was replaced by adenine and p.Gly212Arg means that 212 amino acid glycine was changed to arginine].
The mutations were then confirmed by Sanger’s method. The c.634G>A mutation in the IFT140 gene has been described in individuals with Mainzer-Saldino syndrome (MSS) and the c.2176C>G variant had not been previously described. As compound heterozygosity in IFT140 has not been associated with SLS, a subsequent skeletal survey in her identified flattened femoral heads with widened necks as well as cone-shaped phalangeal epiphyses (Figure 1F), revising her phenotype to Mainzer-Saldino Syndrome (MSS) [Figure 1]. MSS, or conorenal syndrome (CRS), is a rare autosomal recessive disease defined by
- Short stature
- Chronic kidney disease
- Retinal dystrophy
- Phalangeal cone-shaped epiphyses (PCSE)
- Radiographic abnormality of the proximal femur
- Cerebellar ataxia
- Occasionally hepatic fibrosis

Figure 1 from Forbes et al, Am J Hum Genetics, 2018
Extraction of iPSCs from proband’s fibroblasts and gene correction
A One-step protocol was used to generate clonal uncorrected and gene-corrected iPSC lines from the proband’s skin fibroblasts. This protocol combines both reprogramming strategy and CRISPR/Cas9-mediated gene editing. [Figure 2 A]
The Cas9 repair template included correction of the c.634G>A variant and a synonymous 3 bp change to facilitate the identification of corrected clones. [figure 2 B]. Further experiments were carried on one uncorrected proband (PR) and another gene-corrected clone (GC) after confirming pluripotency.
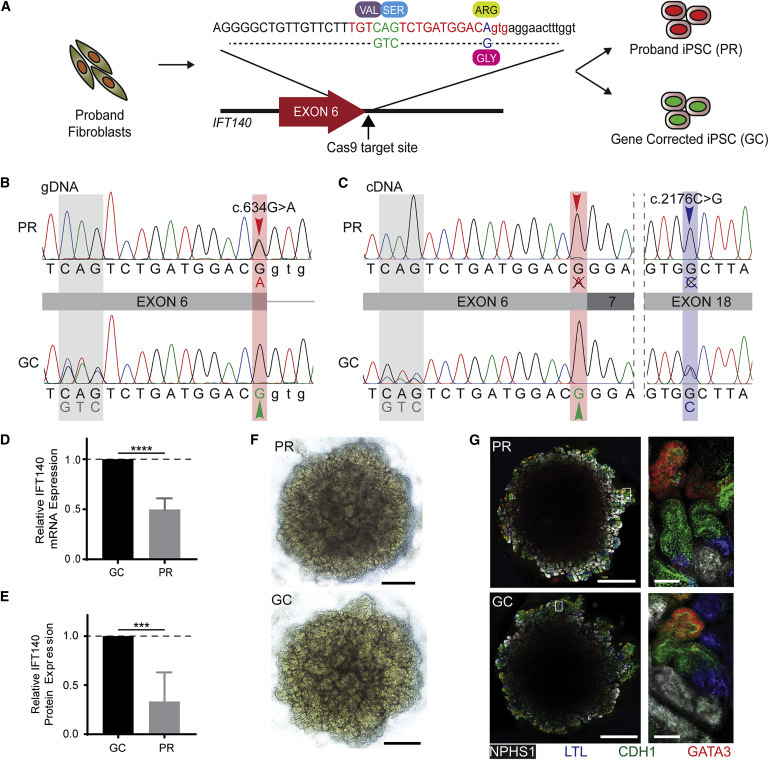
Figure 2 from Forbes et al, Am J Hum Genetics, 2018
Transcriptional analysis
IFT140 transcripts (mRNA) were amplified and sequenced from GC and PR iPSCs. Only the c.2176C>G allele was detectable at the transcriptional level in PR iPSCs (Figure 2C), indicating that the c.634G>A variant results in defective splicing and the incorrectly spliced transcripts degraded. Hence, only one IFT140 allele is transcribed and translated. In contrast, transcription from both alleles was detected in GC iPSCs, showing that gene correction of iPSCs rescues unstable mRNA transcript. This shows that c.634G>A mutation reduces IFT140 mRNA expression by half and protein production by two-thirds (Figures 2D and 2E).
Recreating the disease in the dish
The multicellular kidney organoids differentiated from both PR and GC iPSC lines (Figure 2F). The protocol is described here. Primary cilia stained with acetylated tubulin and ARL13B were identified in the tubular epithelium by immunofluorescence (Figures 3A). Majority of PR cilia showed a clubbed morphology (59%; 95% CI = 54% – 63%), whereas a majority of GC cilia (60%; 95% CI = 56% – 65%) demonstrated wild-type morphology (Figure 3B). Also, PR cilia were significantly shorter than GC cilia (Figure 3C).
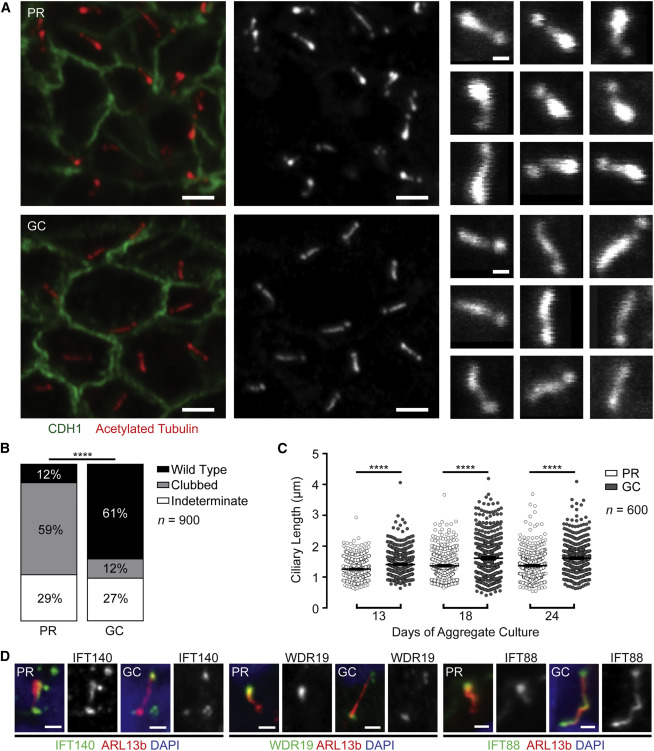
Figure 3 from Forbes et al, Am J Hum Genetics, 2018
Unraveling the basis of Ciliary Dysfunction
As epithelial dysfunction is the proposed cause of renal ciliopathy, the authors isolated and examined the epithelial component of organoids. They performed RNA sequencing of the epithelial subfraction. In order to accomplish this, the group dissociated organoids followed by magnetic bead sorting with EPCAM antibodies (figure 4A). Gene enrichment analysis of genes with significant differential gene expression (p-value < 0.01, n = 954), revealed disturbances in cell adhesion and cytoskeletal interaction pathways (Figure 4B). These findings recapitulated the dysfunctional cellular processes associated with IFT-related ciliopathy.
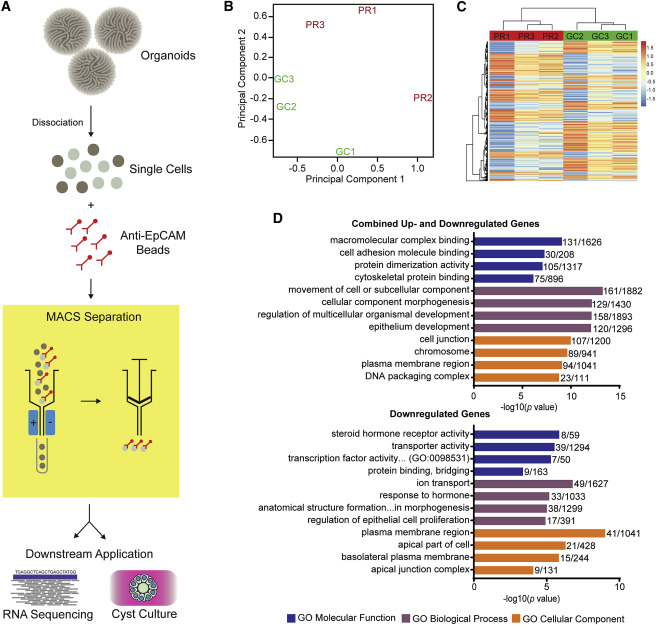
Figure 4 from Forbes et al, Am J Hum Genetics, 2018
Evaluating Cell polarity using spheroid culture
Magnetic bead sorted EPCAM+ cells from both PR and GC organoids were subjected to spheroid culture and stained with markers of apical junctions and apicobasal polarity (Figure 5C). Fewer PR EPCAM+ cells than GC EPCAM+ cells developed spheroids with polarized epithelium ; indicating disturbed cell – cell interactions. Figure 5D). Cilia per nucleus were lower in PR cysts than in GC cysts and were also more variable .
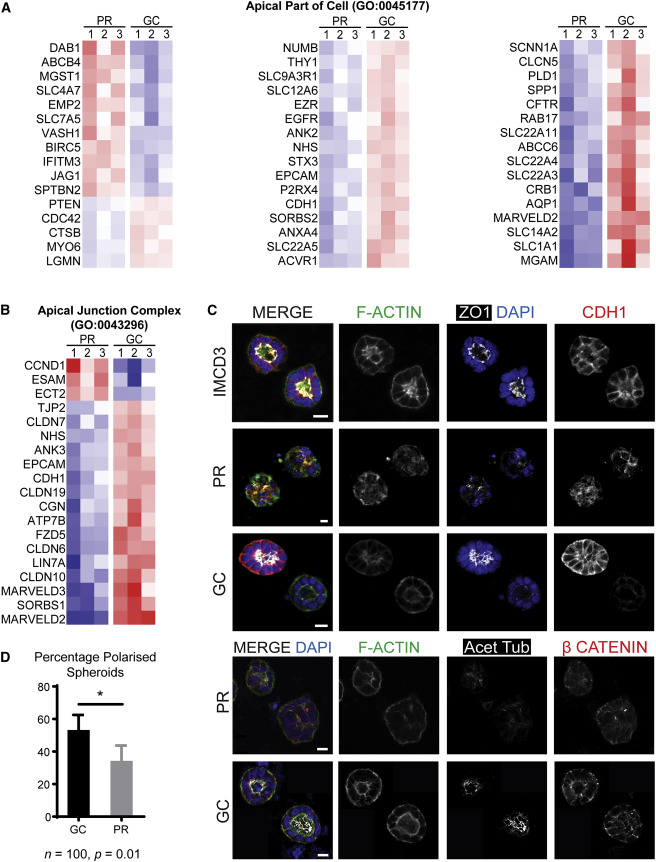
Figure 5 from Forbes et al, Am J Hum Genetics, 2018
Discussion
This first-of-its-kind study demonstrates the utility of proband iPSC derived kidney organoids for functional disease modeling. This approach includes not only the effect of candidate variant but also allows for contribution from the gene modifiers to the phenotype, allowing multigene disorders to be detected. Further, the demonstration of restoration of renal ciliary morphology by gene correction of a single mutated allele supports the efficacy of this approach for validation of novel Variant of Unknown Significance (VUS). The ability to fractionate different cell types from the multicellular 3D disease model (eg. epithelial cells were isolated here as ciliopathies involve epithelial dysfunctional) allows examination of different cell types and their contribution to the disease pathobiology. This can be extrapolated to genetic tubulopathies and glomerular diseases. However as this disease model most closely represents first trimester fetal kidneys, diseases with antenatal or infantile onset (eg. congenital nephrotic syndrome, ARPKD) are more readily studied than those with adult onset (ADPKD).
Conclusion
This study demonstrates that with proband’s cells derived kidney organoid it is possible to recapitulate the disease phenotype in the dish, and its rectification after gene correction helps validate the genetic causality. This opens new avenues for precision medicine by providing personalized screening models and validation of proposed therapies. The authors deserve accolades for their impressive work and especially for the coordinated clinical and laboratory team approach making this possible. It will be intriguing to see replication of this approach in other kidney diseases. Apart from this, there are other perks of having kidney organoids - watch this amazing video by author Melissa H. Little and read this article.
What will be the applications of this discovery in your clinical practice? Which patients will benefit the most? What are the challenges for its implementation? Come and discuss these questions and many more at our Twitter journal club, #NephJC.
Summary by Divya Bajpai, Nephrologist, Mumbai and NSMC Intern, Class of 2018
Tom Forbes (@Kidney_Tom), one of the authors from this week’s NephJC chat on using stem cells to make organelles to serve as patient specific disease models gives his thoughts on the field and where this study fits.