
#NephJC Chat
Tuesday October 11 at 9 pm Eastern
Wednesday October 12 at 9 pm IST
Nature Communications 2022 Aug 15;13(1):4765. doi: 10.1038/s41467-022-32543-2.
PKD1 and PKD2 mRNA cis-inhibition drives polycystic kidney disease progression
Ronak Lakhia, Harini Ramalingam, Chun-Mien Chang, Patricia Cobo-Stark, Laurence Biggers, Andrea Flaten, Jesus Alvarez, Tania Valencia, Darren P Wallace, Edmund C Lee, Vishal Patel
PMID: 35965273







Introduction
ADPKD is the most common inherited condition and the fourth leading cause of kidney disease, affecting 12 million people worldwide (i.e., between 1 in 500 and 1 in 1000 people). Patients with ADPKD lose between 2 and 7 ml/min/1.73 m2 of GFR per year and most individuals reach ESKD before age 60, but can vary from before age 40 to after age 70.
What do we know about ADPKD mutations and pathophysiology so far?
ADPKD is caused by mutations in the genes encoding polycystin 1 (~60-80% of cases; gene: PKD1; protein: PC1) and polycystin 2 (15-30% of cases; gene: PKD2; protein: PC2). These polycystins are cell membrane proteins that play a major role in calcium intracellular signaling and JAK/STAT pathway regulation, though the breadth of their role in the human body is poorly understood. A few other genes -GANAB Borath et al, 2016, DNAHB1 Vinh T Huynh et al, 2020 and IFT140 Senum et al, 2021 have been identified in families with polycystic kidney disease.
ADPKD develops when the expression of PKD1 or PKD2 is reduced by more than 70-80% (the gene dosage theory). ADPKD pathogenesis is classically explained using a two-hit model, where the germline PKD1 or PKD2 mutation provides the first hit, and cyst formation follows a second somatic (acquired) mutation in the kidney tubular cells (Figure 1). In line with this theory, in 2018, Tan et al. performed exome sequencing on 65 cysts from 9 ADPKD patients and identified acquired PKD1/PKD2 mutations in ≥90% of cysts. Effectively, these cyst-forming tubular cells with a germline PKD gene first hit and a somatic PKD gene second hit would be predicted to have markedly reduced or absent PKD expression and thus develop ADPKD. The timing of the somatic mutation impacts the onset and the growth rate of cysts.

Figure 1. Two-hit model in ADPKD pathogenesis. From Saigusa and Bell, 2015.
There are a few reported cases of individuals with ADPKD who inherited more than one PKD gene mutation in their germline and effectively have received both hits before they are born (Durkie et al. 2020), though this is exceedingly rare as most biallelic PKD mutations are lethal to the embryo. This is possible in the presence of hypomorphic allele(s) yielding a compound heterozygote or a bi-allelic homozygote with at least one weak PKD1 or PKD2 hypomorphic allele (that is, with a reduced level of activity or expression. Figure 2 from Bergmann et al. 2018). The hypomorphic variants produce various levels of severity of the disease owing to a differential decrease in the final protein generated. Two hypomorphic variants are shown in Figure 2. The PKD1-Y528C allele results in a milder phenotype similar to that in patients with mutations in PKD2, whereas the PKD1-R3277C allele can result in a phenotype that ranges in severity from just a few cysts to adult-onset disease to early-onset disease, depending on which PKD1 allele is present in trans.

Figure 2. PKD severity by genotype, including the roles of hypomorphic and fully penetrant allele genotypes. The noted modifying factors have a marked impact on disease severity. Figure from Bergmann et al. 2018.
A proposed mechanism of disease progression in ADPKD relates to the snowball effect, where local factors secreted by cysts promote cystogenesis in adjacent tubules. This phenomenon was first described in a 2015 paper by Leonhard et al. The authors depleted Pkd1 in a small number of mice kidney cells and noted that cysts tended to form in clusters near the Pkd1-deficient cells. It was shown that cyst-forming tubules influence neighboring healthy cells to adopt a PKD-like phenotype through paracrine signaling. As shown in Figure 2, the cumulative kidney damage that occurs in ADPKD also promotes further cyst progression through tubular obstruction and regional ischemia. Figure 3 – borrowed from Lanktree et al., 2018 – summarizes the factors known to play a role in the pathogenesis of ADPKD.
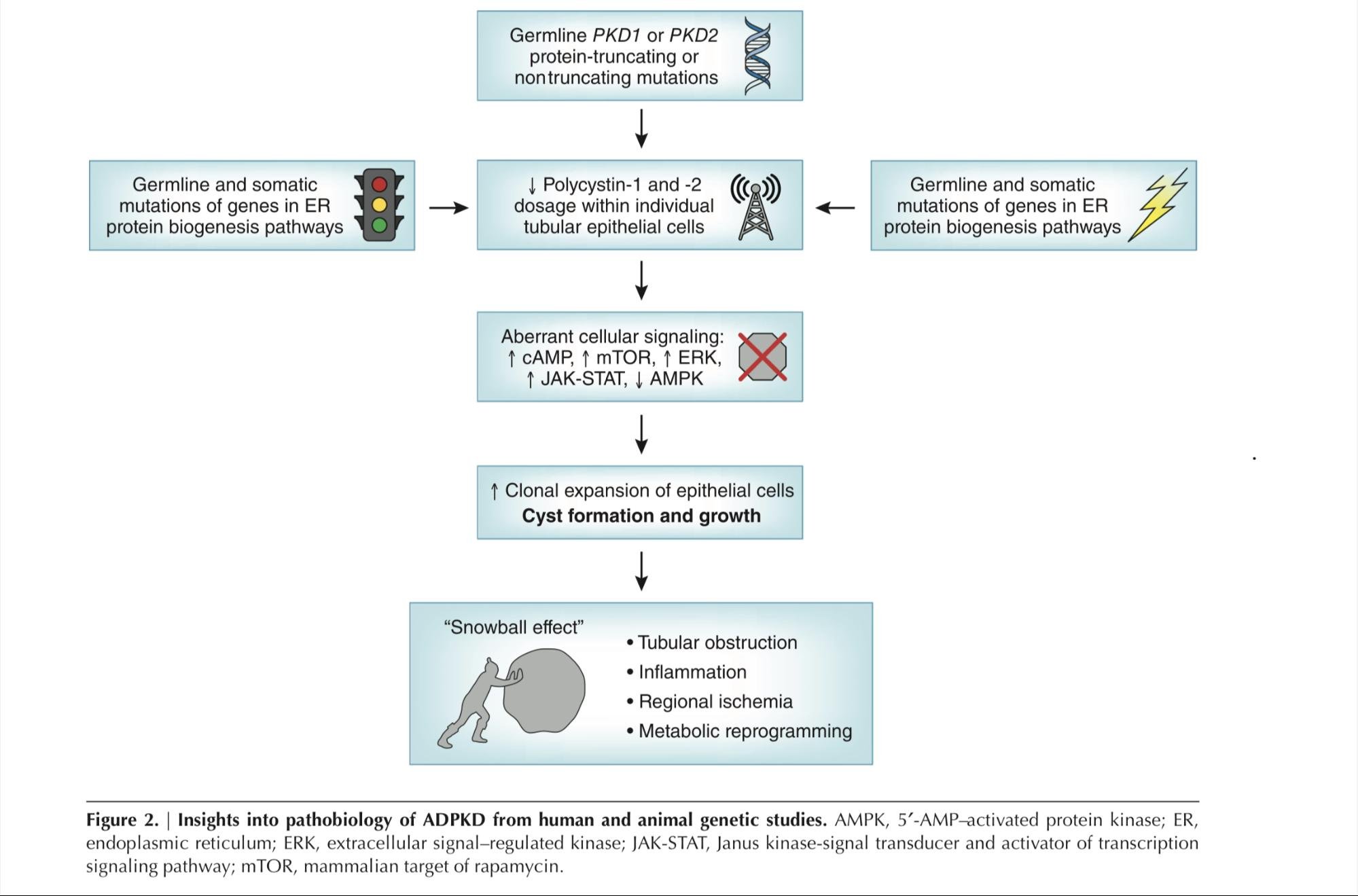
Figure 3: Insights into Autosomal Dominant Polycystic Kidney Disease from Genetic Studies. From Lanktree et al., 2018.
Challenges in the management of ADPKD:
ADPKD is a disease of insidious onset and by the time serum creatinine is elevated, a significant portion of the normal kidney parenchyma has already been replaced by cysts. Most patients with ADPKD develop ESKD in the later part of their lives (median age 58), though early onset ADPKD in childhood and adolescence can also occur. Additionally, there are noted extrarenal manifestations with early onset. For example, nearly 20% of children with asymptomatic ADPKD have hypertension.
Current management of ADPKD is inadequate. Most patients are recommended to maintain a high fluid intake (≥3L per day) in order to suppress vasopressin signaling, a potentiator of cyst growth. In patients at high risk of progression (e.g., high Mayo classification score or high ProPKD score), the vasopressin receptor antagonist tolvaptan can be prescribed. However, there is no treatment to prevent cyst formation in asymptomatic individuals with known PKD1 or PKD2 gene mutations, or those with early disease. There have been attempts to artificially increase PKD1/2 gene expression; however, this comes with the risk of over-expressing these genes to supra-physiologic levels, which also promotes cystogenesis as demonstrated by Thivierge et al. in 2006. It seems the polycystins are sort of like Goldilocks: their levels need to be just right in order to maintain kidney health and avoid cysts. While the idea of preventing kidney cysts in ADPKD is a tantalizing proposition, there are currently no preventive options for ADPKD.
As mentioned, ADPKD is considered a dominant disease in that individuals most often carry a single germline mutation in PKD1 or PKD2. In some cases, it is a truncating variant (no protein is produced), and in other cases it is a hypomorphic variant (a protein with reduced function is produced). In either case, patients still have one healthy copy of the gene. This begs the question: could boosting the levels of hypomorphic or non-mutated copies be a treatment strategy? We discuss a groundbreaking research paper by Dr. Ronak Lakhia and colleagues who, through their series of studies, have uncovered a potential therapeutic target that seems to do just this.
In brief, Lahkia et al. show that microRNA 17 (miR-17) binding to a distal site in the PKD1 and PKD2 genes represses their expression, and that inhibiting miR-17 effectively rescues (or de-represses) polycystin levels. miR17 is normally produced at low basal levels in healthy adult kidneys, but it is known to be over-expressed in ADPKD. Using in vitro mouse and human ADPKD cell models as well as in vivo mouse models, they show how anti-miR-17 treatment has the potential to treat ADPKD along the continuum of disease: from prevention of cyst formation in ADPKD altogether in sub-clinical cases to attenuation of cyst growth in established disease.
The study
Methods
In their paper, the authors used modified mouse- and human-derived cells, as well as live mouse models to explore the role of miR-17 in PKD pathogenesis.
Mouse models and cell lines used
Mouse PKD cell lines
The authors use the Pkd1RC/- cellular mouse ADPKD model, which has one germline hypomorphic variant (the mouse version of R3277C; shortened to RC for simplicity) and one truncating variant. This mimics the In utero onset genotype from Figure 2. They compare these with the Pkd1RC/+ cell line, which has one copy of the RC hypomorphic variant alongside a “floxed” allele. Floxed alleles contain specific sequences that enable genetic deletion upon addition of an external agent – usually Cre recombinase. Essentially, floxed alleles function as healthy copies of the gene unless this external agent is added, in which case the gene is deleted. This system allows researchers flexibility to edit the genomes of the cell lines or mouse models they are using at desired timepoints in their experiments.
PKD mice models
The authors used Pkd1RC/RC mice, which have the RC genetic change in both of their Pkd1 copies. This recapitulates the adult onset ADPKD genotype described in Figure 2. These mice were crossed with mice containing Pkd1 floxed alleles, yielding progeny that had one copy of hypomorphic Pkd1-RC and one copy of floxed Pkd1 (Pkd1F) that could then be removed by addition of an external agent. This allowed the authors to generate mice that had intact and deleted Pkd1 alleles, depending on whether Cre recombinase was added: Pkd1RC/F (Pkd1RC/+) and KspCre+;Pkd1RC/F (Pkd1RC/-), respectively.
Human PKD cell lines used
Cell lines were derived from human PKD cyst cells from discarded nephrectomy tissue. PKD1 and PKD2 mutation analysis was done to confirm the genotype for these samples.
Methods used to inhibit the effects of miR-17
Deletion of the miR-17 binding site in cell lines and live mice
The miR-17 binding motif in Pkd1 and Pkd2 is in the 3’ untranslated region (3’ UTR) of these genes, called the mir-17 binding element or MBE. The authors used CRISPR-Cas9 technology to remove the MBE in live mice and in cell lines, which are annotated with the ∆17 designation throughout the text (method shown in Figure 4). They performed this in mice and cells with and without PKD truncating mutations, which produced for example cells where one allele has RC variant and MBE deletion and the other allele has a truncating allele (Pkd1RC∆17/-), or cells where the Pkd1 allele was the classic Pkd1RC/- while the MBE was deleted from Pkd2 (Pkd1RC/-; Pkd2∆17/∆17 cell line).

Figure 4. Method used to excise miR-17 binding element from Pkd1 mRNA.
Addition of a miR-17 inhibitor (RGLS4326)
The authors developed a short oligonucleotide inhibitor of miR-17, which they first described in 2019. The effect of adding this inhibitor was tested in both cell lines and mouse models with two important controls in each case: a vehicle control (no oligonucleotide) and a control oligonucleotide (a random oligonucleotide that should not inhibit miR-17, used to assess whether adding an exogenous nucleotide sequence affects the cell/body).
For the mouse models, the authors studied the effect of injecting this inhibitor at 3 different timepoints to model the effect of inhibiting miR-17 at different stages of ADPKD disease:
Cyst prevention study: The mice were injected on postnatal days (P) 10, P11, P12, and P16 and sacrificed on P18. Nontransgenic strain-matched control mice were sacrificed on the same days.
Disease stabilization study: The mice were injected on P16 and P17 and sacrificed at P26. Of note, one mouse from the study succumbed to the disease and died earlier than 26 days of age.
Long-term Cyst Treatment study: mice were injected on P16 and P17, and then injected again with RGLS4326 either weekly or biweekly until 18 weeks of age.
Results
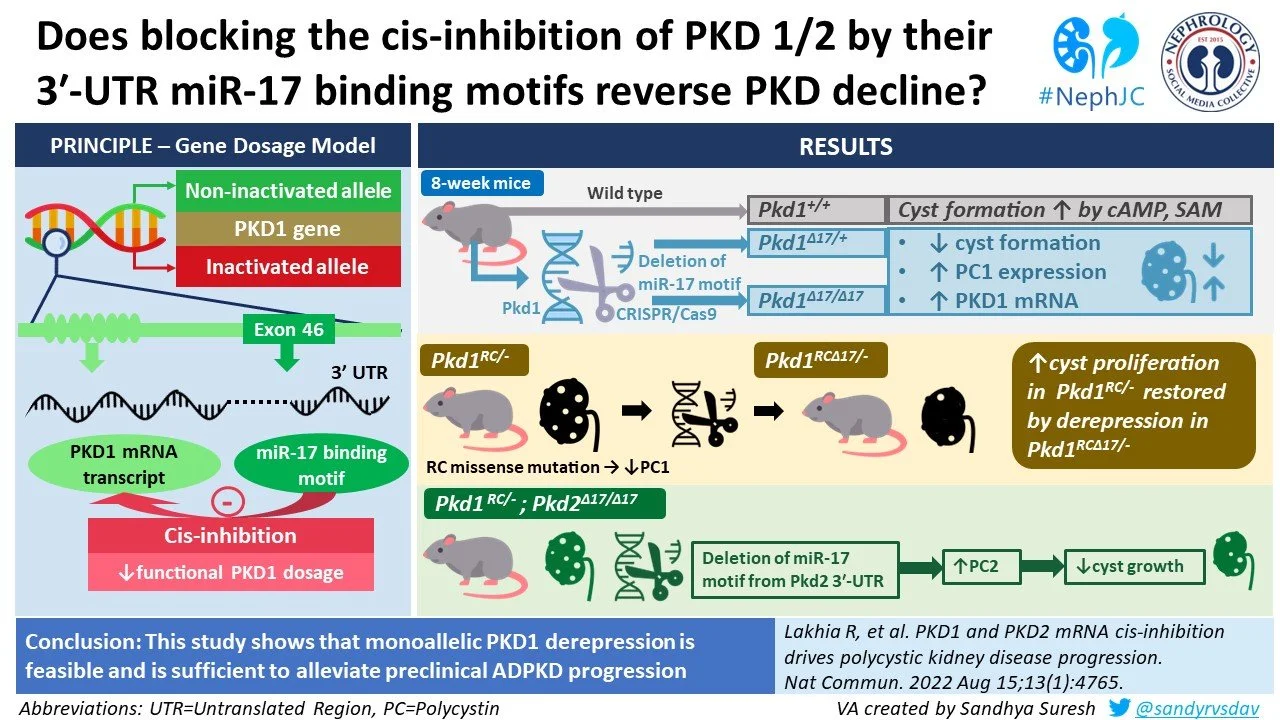
PKD1 is cis-repressed via its 3’ UTR miR 17 binding motif
The investigators started off with studying the effect of deleting the miR-17 binding element (MBE) in normal mouse kidneys. Since miR17 is expressed at very low levels beyond the embryonic period in healthy mice, this first part of the paper was conducted in embryonic mice. To recap from the Methods section, we described how the mRNA of the Pkd1 gene has a miR-17 binding element (MBE) in the 3’ UnTranslated Region (UTR) segment, and that binding of miR-17 to the Pkd1 mRNA prevents translation of PC1. The MBE was targeted by sgRNA and using CRISPR/Cas9 editing, a Pkd1 allele lacking miR-17 binding site (Pkd1∆17) was developed.
Pkd1∆17/∆17 mice were notably found to have normal expression of PC1 despite the absence of UTR-miR 17 binding motif even at 18 weeks of age, hence proving that miR-17 binding motif deletion does not affect PC1 expression (Figure 5). Human healthy kidneys have a diminished output of miR-17 as they mature. This decrement correlates with the lack of cis-inhibition of Pkd1 due to low basal activity of miR-17.

Figure 5. Removal of the miR-17 MBE in Pkd1 mRNA does not affect PC1 levels in healthy mice.
Then, they exposed mice with and without deletion of miR-17 MBE deletion to cystogenic compounds cAMP and S-adenosylmethionine (SAM). As expected, they saw that wild type mice with two normal Pkd1 alleles (Pkd1+/+) responded by forming cysts. In contrast, cystogenesis was attenuated in mice with deletion of the MBE in one or both alleles (Pkd1∆17/+ and Pkd1∆17/∆17, respectively; Figure 6g). As expected, in these cytogenetic conditions in the developing kidney, a higher expression of PC1 was noted in Pkd1∆17/+ and Pkd1∆17/∆17 compared to Pkd1+/+ cells (Figure 6h).

Figure 6. Effect of miR-17 binding element (MBE) deletion on response to cystogenic stimuli. Cyst formation was reduced (g) and PC1 levels were lower (h) in mice with MBE deletion.
When the relative amount of wild type vs. ∆17 mRNA transcripts were quantified by qRT-PCR method, they observed that the ∆17 allele contributed nearly 50% more transcripts than its wildtype counterpart. These observations suggest that the wild type Pkd1 mRNA is inhibited by miR-17 but its inhibition is evaded in Pkd1∆17 mRNAs in embryonic kidneys.
Endogenous monoallelic Pkd1 derepression alleviates polycystic kidney disease
While miR-17 is lowly expressed in healthy adult kidneys, it is known to be overexpressed in ADPKD. Therefore, the investigators then studied whether repression of Pkd1 by MBE also occurs in ADPKD, and whether the inhibition of miR-17 had an impact on the disease.
They first studied the Pkd1RC/− cellular ADPKD model, where the RC allele is a hypomorphic variant that reduces PC1 levels (see details in the Methods). Similar to what was seen in the embryonic mice with wildtype Pkd1, elimination of miR-17 motif from the Pkd1RC allele reduced the cystogenic effect cAMP and SAM (Figure 7).

Figure 7. A higher proliferation rate and 3D cyst size was noted in Pkd1RC/- cells compared to Pkd1RC/+ cells, which was normalized in cells with the miR-17 MBE was removed (Pkd1RC∆17/- cells).
The research team then studied the effect of deleting of 3′-UTR ∆17 in vivo. They had CRISPR-edited fertilized eggs from an ADPKD mouse model (Ksp-Cre;Pkd1RC/RC; see Methods for details) to eliminate the miR-17 motif. They then implanted these eggs into pseudo-pregnant surrogate female mice and obtained progeny mice that have Ksp Cre; Pkd1RC∆17/RC genotypes. They repeated this procedure twice in order to develop 3 separate colonies of founder mice. This strategy is an alternative to using littermates from one mouse colony, which is touted to increase the confidence in results by deriving separate biological replicates.
The team observed:
Whereas Pkd1RC/- mice developed cysts, fairly normal histology and function was noted in Pkd1RC∆17/- mice at 18 days (Figure 8).

Figure 8. Deletion of the Pkd1 MBE in mice with Pkd1 mutations mitigates the development of ADPKD phenotypes.
For each of their 3 founder progeny, a reduced PC1 expression, severe cystic kidney disease, an increased kidney-weight-to-body-weight (KW/BW) ratio, and higher serum BUN levels were observed in Pkd1RC/- mice compared to Pkd1RC/+ mice were observed. KW/BW and serum BUN were nearly normalized in Pkd1RC∆17/-mice compared to Pkd1RC/- mice (Figure 9).

Figure 9. Deletion of the Pkd1 MBE in mice with Pkd1 mutations improves KW/BW ratio and BUN.
Pkd1RC∆17/-mice had improved long-term outcomes compared to Pkd1RC/- mice, including lower cyst burden and better survival at 8 weeks (Figure 10).

Figure 10. Deletion of the Pkd1 MBE in mice with Pkd1 mutations improves survival at 8 weeks.
Then, the authors performed bulk RNA sequence (RNA-Seq) analysis on kidney samples from 18 day old Pkd1RC/+, Pkd1RC∆17/+, Pkd1RC/-, and Pkd1RC∆17/- mice. They noted dysregulation of an extensive network of gene transcripts in cystic Pkd1RC/- kidneys compared to non cystic Pkd1RC/+ control kidneys, including significant upregulation of 4157 and downregulation of 2067 mRNAs (Figure 11). Impressively, >95% of dysregulated mRNAs showed improved (or normalized) expression in Pkd1RC∆17/- kidneys .

Figure 11. Deletion of the MBE in mice with Pkd1 mutations restores a normal gene expression profile in RNA sequencing experiments.
Preventing Pkd2 cis-inhibition attenuates cyst growth in Pkd1-mutant models: Can PC2 compensate for PC1?
The work so far has focused on the role of the miR-17 binding motif in Pkd1 in ADPKD pathogenesis. However, Pkd2 also contains this domain. Therefore, the authors studied whether deleting this domain in Pkd2 could rescue the Pkd1-depleted phenotype in ADPKD. They used CRISPR/Cas9 in their ADPKD cell model to delete the miR17 MBE from Pkd2 and yield Pkd1RC/-; Pkd2∆17/∆17cells. They showed that these cells had higher Pkd2 mRNA and PC2 protein levels compared to their unedited parental Pkd1RC/- cells, while PC1 expression remained unchanged. PC2 ∆17 was associated with decreased cystogenesis and cyst growth upon treatment with pro-cystogenic stimuli in these cell culture models. Remarkably, removal of the miR17 MBE in Pkd2 also drastically improved cyst production in vivo in mice with the Pkd1RC/- genotype (Figure 12) .

Figure 12. Deletion of the Pkd2 MBE in mice with Pkd1 mutations reduces kidney volume and normalizes protein markers associated with cystogenesis in ADPKD.
Acute blockade of Pkd1 and Pkd2 cis-inhibition ameliorates PKD
The investigators used an anti-miR-17 oligonucleotide that they had previously characterized, RGLS4326, as a tool to acutely block Pkd1 and Pkd2 cis-inhibition. As expected, administration of RGLS4326 to mice with Pkd1RC/- led to an increased expression of PC1 and PC2 as compared to control oligonucleotides. When RGLS4326 was administered to mice with Pkd1 or Pkd2 mutations and genetic deletion of the miR17 binding motif, there was no difference in PC1 and PC2 levels respectively. This confirms that the upregulation of polycystins by this oligonucleotide works as expected: it relies on the miR-17 motif in Pkd1/2 3′-UTRs.
Next, the authors assessed whether administering RGLS4326 to mice with Pkd1 mutations reduced cyst formation, similar to elimination of the miR-17 binding motif. In a first experiment, RGLS4326 was administered over 4 days (P10, 11, 12, 16) and kidney histology was quantified at 18 days post-natal (Figure 13d). Treatment with RGLS4326 was associated with less cystogenesis at both timepoints, as well as with lower relative kidney weight (KW/BW), lower BUN and lower creatinine (Figure 13 e-g). These improvements persisted at 26 days post-natal (Figure 13 h-k). The authors then assessed whether treatment at weekly and bi-monthly intervals improved long-term outcomes. They saw that, in both dosing regimens, cystogenesis was markedly reduced with treatment (Figure 13l). Mouse survival was also significantly longer at 18 weeks (Figure 13m).

Figure 13. Anti-miR17 oligonucleotide treatment improves cystogenesis in mice with Pkd1 mutations in the short (P18), medium (P26) and long term (P125).
PKD1 ∆17 or PKD2 ∆17 alleles reduce cyst growth of patient-derived primary ADPKD cultures
Finally, the team assessed whether this miR17 mechanism they characterized in mice was relevant in humans. They derived human kidney cell lines from four ADPKD affected patients. Cell lines #1, #3 and #4 had heterozygous PKD1 mutations while cell line #2 had a heterozygous PKD2 truncating mutation and a missense PKD1 heterozygous mutation. Once again, they used the CRISPR-Cas9 system to remove the miR-17 MBE from these cells. This produced higher PC1 levels in all four PKD1∆17 cultures compared to their respective mock-transfected parental controls. In cell culture, the PKD1∆17 and PKD2∆17 cells formed smaller cysts and exhibited lower proliferation rates (Figure 14).
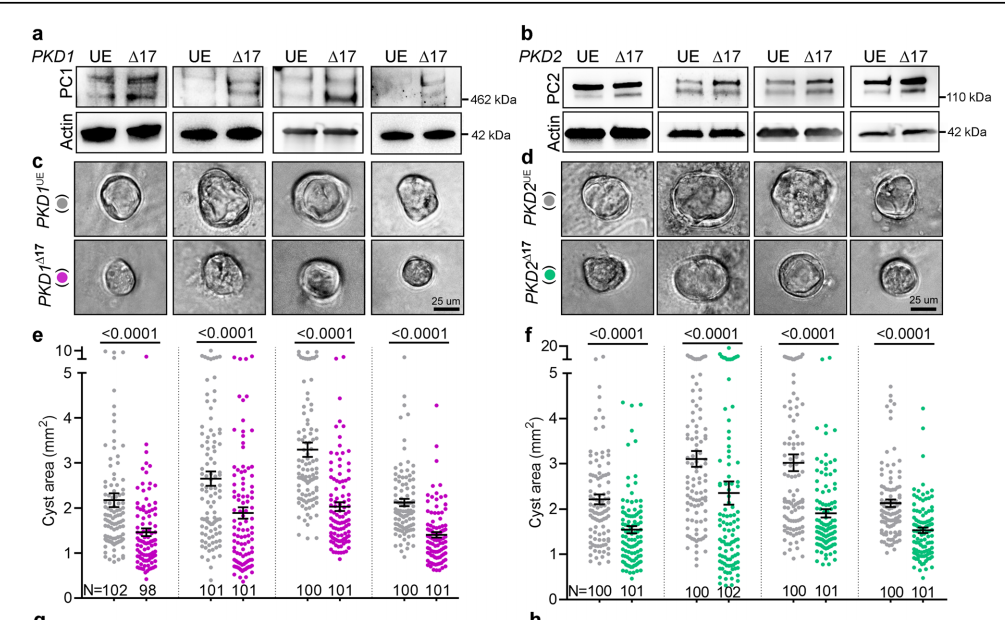
Figure 14. Genetic deletion of the miR-17 binding domain in ADPKD patient-derived cell lines improved PC1 / PC2 levels and reduced cyst-forming ability.
Summary
Altogether, this groundbreaking work by Lakhia and colleagues establishes miR-17 as a novel important regulator of ADPKD that may be therapeutically targeted. They show that polycystin protein levels can be repleted in mice with PKD mutations and in human cell lines derived from ADPKD patients by deleting the Pkd1 3’UTR- miR-17 binding motif or by blocking miR-17 function using an oligonucleotide inhibitor (RGLS4326). miR-17 normally represses Pkd1 and Pkd2 in ADPKD, and “de-repression” of the polycystins might thus constitute a viable treatment option for this disease.
To understand this better, imagine a reading lamp that has a power switch to turn it on or off. The miR-17 binding motif is the switch to allow PKD1 expression to turn on and off. If the switch is removed (or the miR-17 binding site is deleted) the lamp remains always lit. Alternatively, an inhibitor (RGLS4326) can prevent the light from being switched off, leaving the lamp to produce light (the good unmutated copy of the polycystin 1 protein continues to produce protein.).
Discussion
The most common cause of ADPKD is a heterozygous loss of function mutations involving the PKD1 gene. Despite having at least one healthy copy of the PKD1 gene at birth, many individuals develop kidney cysts and kidney failure. It was previously known that somatic mutations that lead to total loss of PKD1 expression promote cystogenesis, and that disease progression follows a vicious cycle where existing cysts and kidney dysfunction promote the development of more cysts and progression of kidney failure (Figure 3). These novel findings by Lahkia et al. describe the role that miR-17 plays in dampening polycystin protein production, and how inhibiting this microRNA can boost the expression of hypomorphic or healthy copies of PKD1 to mitigate cystogenesis and disease progression. The authors inhibit miR-17 function using genetic deletion of its binding site and using an exogenous oligonucleotide miR-17 inhibitor, RGLS4326. In tandem with the work they present in this paper, the authors have also begun piloting RGLS4326 in humans, and in fact showed promising preliminary Phase 1 trial results at KidneyWeek 2021.
Though an unexpected finding, the de-repression of Pkd2 in Pkd1-mutant models has increased stability of the disease. This suggests that the interactions between both the polycystins are significant and supportive. However, Pkd2 de-repression versus Pkd1 de-repression for treatment is debatable. A long-term study may address this issue.
We now understand that treating pre-clinical stages of ADPKD is possible. This warrants a reconsideration of genetic testing in asymptomatic first degree family members of ADPKD patients, including children.
Another fascinating direction of these therapeutic options is the probable effect of this therapy on the extra-renal manifestations of the disease. It is to be seen whether stabilization of polycystin achieved would translate to a systemic benefit. The possibility of combining this therapy with the established standard of therapy viz., tolvaptan has to be studied for efficacy, adverse effects and a head to head comparison is awaited.
The future directions of therapy in ADPKD presented by this study points towards an exciting option of killing the disease before its onset, mitigation of severity of established disease, and slowing the rate of progression. It also opens up avenues to treating other genetic diseases through targeted mRNA de-repression.
Summary prepared by Srikanth Bathini
Nephrologist, Hyderabad, India
NSMC Intern, class of 2022
Reviewed by Kelly Hyndman, Caitlyn Vlasschaert, and Matt Lanktree
Abbreviations used in this review:
ADPKD = Autosomal Dominant Polycystic Kidney Disease
PKD = Human Polycystic kidney disease gene
Pkd = Mouse Polycystic kidney disease gene
PKD1 = Polycystic Kidney Disease 1 gene
PKD2 = Polycystic Kidney Disease 2 gene
PC1 = Polycystin 1
PC2 = Polycystin 2
Pkd1 +/+ = Wild type, Normal gene
Pkd +/hp = Pkd 1 with one normal allele and one hypomorphic allele
Pkd -/- = Pkd 1 double null alleles
Pkd 1 +/- = Pkd1 with one normal allele and one null allele.
Hypomorphic mutations: inactivating mutations or mutations that reduce the function of the gene product (hypomorphic mutations))
PKD1Y528C/+ = Hypomorphic allele that results in a phenotype similar to that in patients with mutations in PKD2
PKD1R3277C/+ = Hypomorphic variant with a phenotype that ranges in severity from just a few cysts to adult-onset disease to early-onset disease, depending on which PKD1 allele is present in trans.
3’ UTR mRNA= 3’ untranslated region of PKD mRNA
MBE = Micro RNA binding element
miR-17 = microRNA 17
3’ UTR miR-17 BE = Binding element (motif) of the3’ Untranslated region of the PKD mRNA.
Pkd1∆17/∆17 = Pkd1 with deletion of miR-17 binding motif of 3’ untranslated region of Pkd1 mRNA in both alleles.
Pkd1∆17/+ = Pkd1 with deletion of miR-17 binding motif of 3’ untranslated region of Pkd1 mRNA in one allele, and an intact, normal (wild type) allele.
Pkd1RC/− = A cellular ADPKD model which is a collecting duct derived (PKD1 variant p.Arg3277Cys (RC) humanized) mouse cell line with the missense RC mutation on one Pkd1 allele and an inactivated other allele. The RC mutation leads to a reduced functional PC1 level (Hypomorphic variant).
Pkd1RC/+ = Missense RC mutation on one allele with a normal allele.
Pkd1RC/RC = Double Missense RC Mutations in both alleles.
Pkd1RC∆17/- = RC mutation with miR- 17 binding motif deleted and a null allele.
KW/BW = Kidney Weight /Body weight ratio
RGLS4326 = anti-miR-17 oligonucleotide
Pkd1 Flox/Flox = Floxed alleles contain specific sequences that enable genetic deletion upon addition of an external agent – usually Cre recombinase. Essentially, floxed alleles function as healthy copies of the gene unless this external agent is added, in which case the gene is deleted. This system allows researchers flexibility to edit the genomes of the cell lines or mouse models they are using at desired timepoints in their experiments.
Px = post-natal day x (x= any given number)