
#NephJC Chat
Tuesday Sept 3, 2019 at 9 pm Eastern Daylight Time
Wednesday Sept 4, 2019 9 pm British Summer Time, 1 PM Pacific Daylight Time
Wednesday Sept 4, 2019 9 pm Indian Standard Time
Cell. 2019 Jul 25;178(3):521-535.e23. doi: 10.1016/j.cell.2019.07.002.
Small Molecule Targets TMED9 and Promotes Lysosomal Degradation to Reverse Proteinopathy.
Dvela-Levitt M, Kost-Alimova M, Emani M, Kohnert E, Thompson R, Sidhom EH, Rivadeneira A, Sahakian N, Roignot J, Papagregoriou G, Montesinos MS, Clark AR, McKinney D, Gutierrez J, Roth M, Ronco L, Elonga E, Carter TA, Gnirke A, Melanson M, Hartland K, Wieder N, Hsu JC, Deltas C, Hughey R, Bleyer AJ, Kmoch S, Živná M, Barešova V, Kota S, Schlondorff J, Heiman M, Alper SL, Wagner F, Weins A, Golub TR, Lander ES, Greka A..
PMID: 31348885 Full Text at Cell ($)


As an introduction, this is an excellent place to start, with Matt Spark’s tweetorial:
Introduction
Proteinopathies are diseases associated with the misfolding of proteins and their intracellular or extracellular accumulation that can result in cellular toxicity and cell death. The most prominent proteinopathies are Retinitis Pigmentosa and Parkinson disease, as well as 20 other rare disorders. Several progressive kidney diseases have been linked to proteinopathies and several of these have been lumped in a group of similar diseases called autosomal dominant tubulointerstitial kidney disease (ADTKD).

Figure from Bleyer et al. Kidney International Sept 2014.
A number of genetic mutations have been identified as causative in several of the ADTKDs and two MUC1 and UMOD are proteinopathies. Thus, this has led to a reclassification of ADTKDs based on the gene that is mutated. Many of these disorders have a subtle phenotype with nonspecific findings on biopsy, no biomarker, and slow progression (median age of ESKD is ~50 years old). You should be suspicious if you encounter a strong familial history of ESKD and biopsy showing tubulointerstitial kidney disease of underlying ADTKDs. We are now in an era where genetic diagnosis is possible. To date, there have been 4 different genes with putative causal mutations in ADTKD: MUC1-Mucin-1, UMOD-Uromodulin (Tam Horsfall), HNF1B-Hepatocyte nuclear factor 1-beta, and REN-Renin. This paper focuses on mutations in MUC1.
Mutations in the MUC1 gene (on chromosome 1q21) have been identified to cause progressive tubulointerstitial kidney disease inherited in an autosomal dominant fashion and thus now termed, MUC-1 kidney disease (MKD). The clinical presentation of patients with MKD is usually progressive chronic kidney disease (CKD) in several family members in an autosomal dominant pattern. Patients usually have hyperuricemia and gout and urinalysis is typically bland. The kidney ultrasound may show medullary cysts, although this finding can be absent. Unfortunately, kidney biopsies are nonspecific and typically show tubulointerstitial fibrosis.
The mutation in MUC1 was first described in this landmark paper in Nature Genetics in 2016. This was a challenging mutation to identify because the MUC1 gene harbors multiple variable number tandem repeat (VNTR)s and was difficult to assay. These VNTRs consist of (20–125) copies of a large repeat unit (60 bases) with very high GC content (>80%). Affected individuals have a single cytosine insertion into one VNTR region within the MUC1 coding region leading to a frameshift mutation, resulting in a new truncated protein (MUC1-fs) without the sequences allowing for insertion into cytoplasm. MUC1-fs accumulates and for some reason after many years triggers the unfolded protein response (UPR) leading to cell toxicity and eventually apoptosis.
Currently, there is no specific therapy for MKD and the underlying molecular mechanism is still being elucidated. The authors of this manuscript describes a potential mechanism by which the cargo protein TMED9 cargo receptor induced intracellular accumulation of MUC-1-fs in the early secretory compartments leading to the activation of the unfolded protein response. They also go on to identify BRD4780, a molecule that releases MUC1-fs from TMED9-enriched compartments and promotes its removal by lysosomes. As such, in this manuscript, Devela-Levitt et al. provide a detailed molecular mechanism for MKD involving misfolded protein entrapment and also identify a novel molecule as a promising lead for the potential treatment of rare proteinopathies.
Methods
In this study, the authors generated three different mouse models to test their hypothesis:
mMuc1wt (+) mouse normal Muc1
hMUC1wt (wt) human normal MUC1
hMUC1fs (fs) human mutant MUC1fs .
These models utilized mice with that harbor either the normal Muc1 from the mouse (called mMuc1wt (+)), the normal allele from the human (called hMUC1wt (wt)). It’s important that the normal human MUC1 gene is expressed in the mouse to ensure that this does not lead to any cellular derangements alone. The final mouse model is of the human mutation as described by the Nature Genetics paper. This is what they hope to recapitulate the human disease of MKD.

They also include 2 other cell culture models.
P cells- are epithelial tubular cells isolated from a kidney specimen obtained from a patients with MKD. Control cells are called N cells and are derived from a kidney specimen obtained from a healthy individual. Both of these cells were immortalized following their isolation.
Organoids- these are differentiated from iPS cells from erythroblasts taken from three different patients with MKD or from their unaffected siblings as controls- to help test hypothesis in cells with more potential for human translation.

They also show that P cells have cytoplasmic localization of MUC1-fs as shown above.
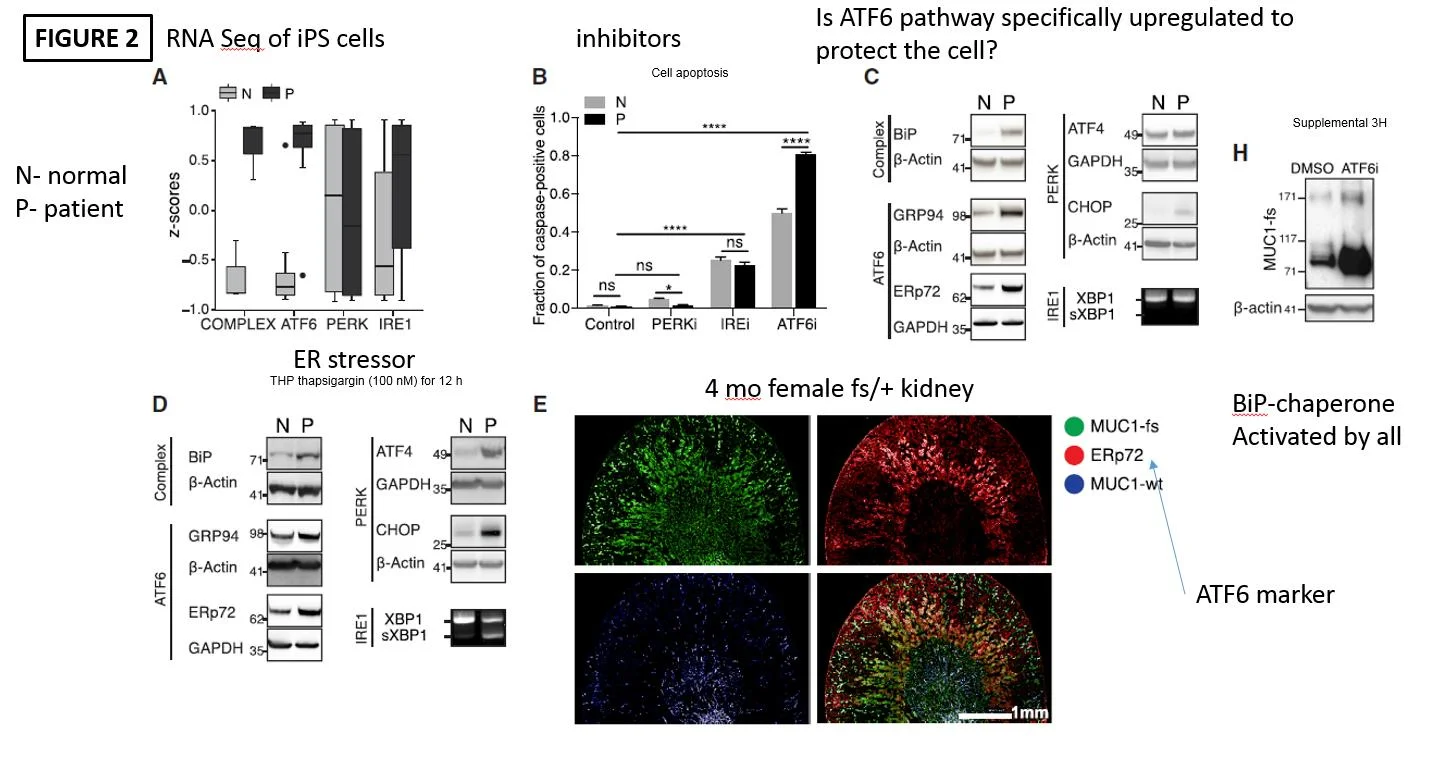
The next step was to understand what exactly is going on with the unfolded protein response (UPR) in MKD. This is a cellular response all cells have to deal with protein debris (misfolded and damaged proteins). This can range from pushing proteins to degratory pathways in lysosomes to apoptosis and cell death. To do this they utilized P and N cells and performed RNA sequencing. As seen in figure 2A they demonstrated enhanced complex and ATF6 pathways in P cells as compared to N cells. This suggests that both of these pathways are enhanced in patients with MUC1 mutations. To review. The ATF6 pathway is one of 3 pathways utilized in the UPR. Potentially, the ATF6 pathway upregulation is a protective mechanism to deal with the MUC1-fs. They went on to give inhibitors of each of the pathways as seen in Figure 2B. The showed that the ATF6 pathway when inhibited resulted in more apoptosis in P compared to N cells. Again, pointing to ATF6 being upregulated and potentially protective in MKD. The rest of the figure goes on to (C) show more evidence of ATF6 pathways upregulated and (D) the application of an ER stressors (thapsigarin) resulted in P cells being more sensitive and finally (E) in the mice with human MUC1-fs having colocaliation with the ATF6 marker ERp72. These findings all point toward an upregulated ATF7 UPR pathway in MKD.

The UPR pathways.
The next step was to identify a compound that would lead to less MUC1-fs accumulation. The investigators developed a high content screen (HCS) using IF-based assay to simultaneously assess the
abundance of MUC1-wt
MUC1-fs
cell number
They used the Broad Repurposing Library, which has a set of 3,713 compounds at different stages of preclinical and clinical development as seen in Figure 3.

The top hit of the screen was BRD4780. This drug was originally developed as an imidazoline receptor ligand, similar to clonidine. It was originally called AGN 192403 and make by Allergan Pharma. It was devoid of all physiologic responses and did not affect BP, intraocular pressure, or induce sedation in animal models.
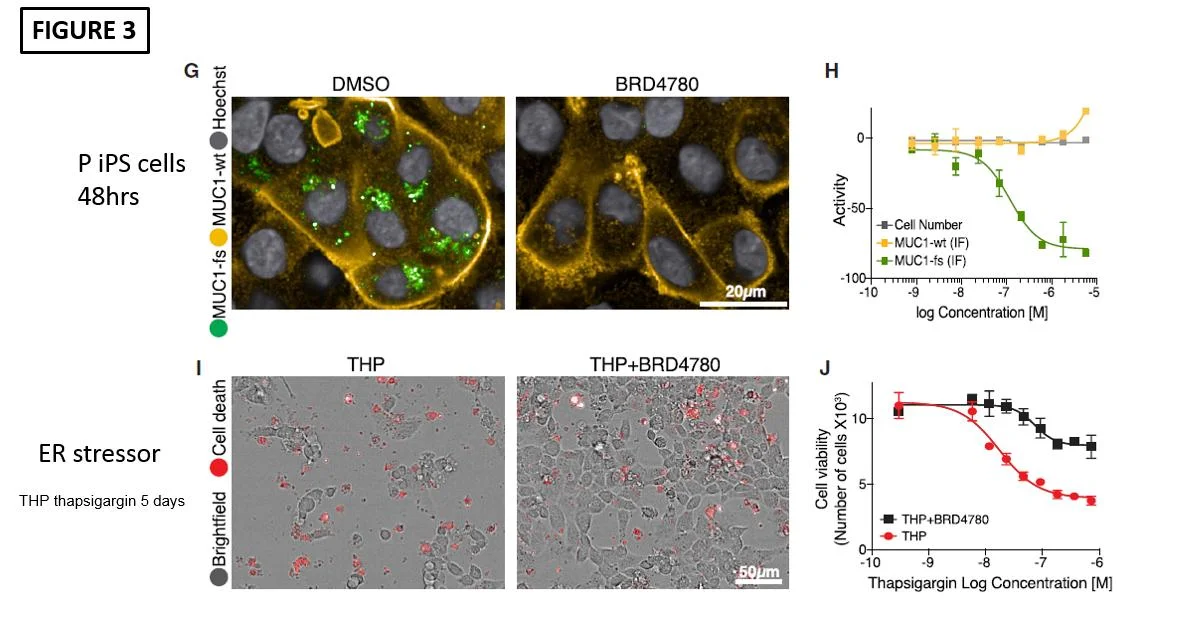
Then the investigators first administered BRD4780 to P cells and found that MUC1-fs indeed was diminished after 48 hours (Figure 3G). In addition, coadministration of the ER stressor, thapsigargin, with BRD4780 resulted in much less cell death (Figure 3I). They also showed that treating mice (via oral gavage) with expression of human MUC1-fs also demonstrated diminished MUC1-fs levels.
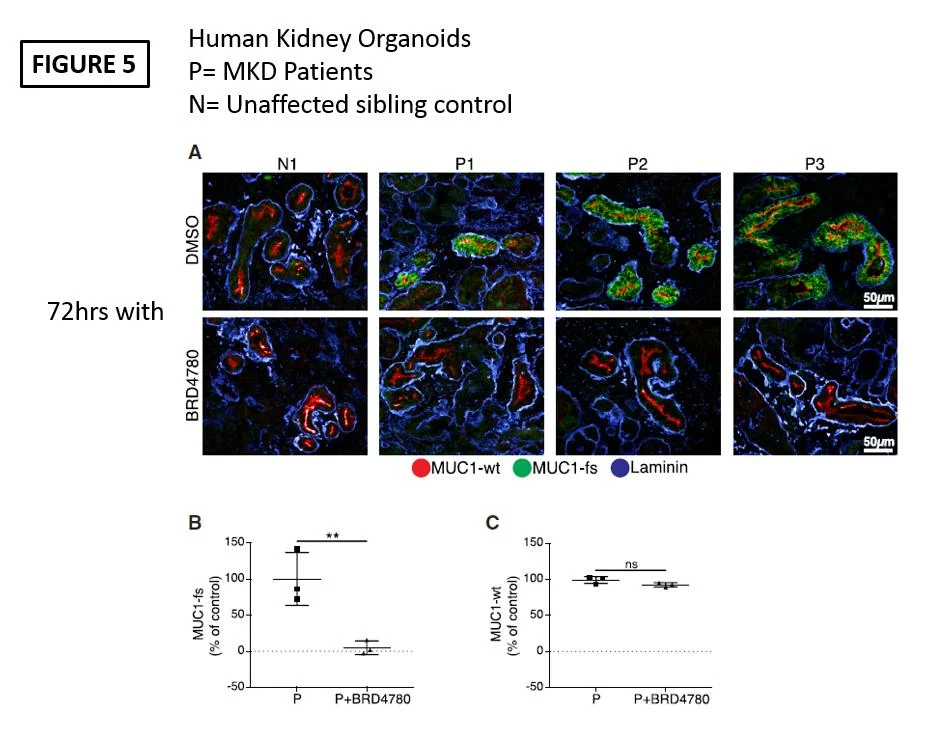
In order to demonstrate human relevance they gave BRD4780 to human organoids derived from P cells and this also showed reduced MUC1-fs after 72 hours of treatment.
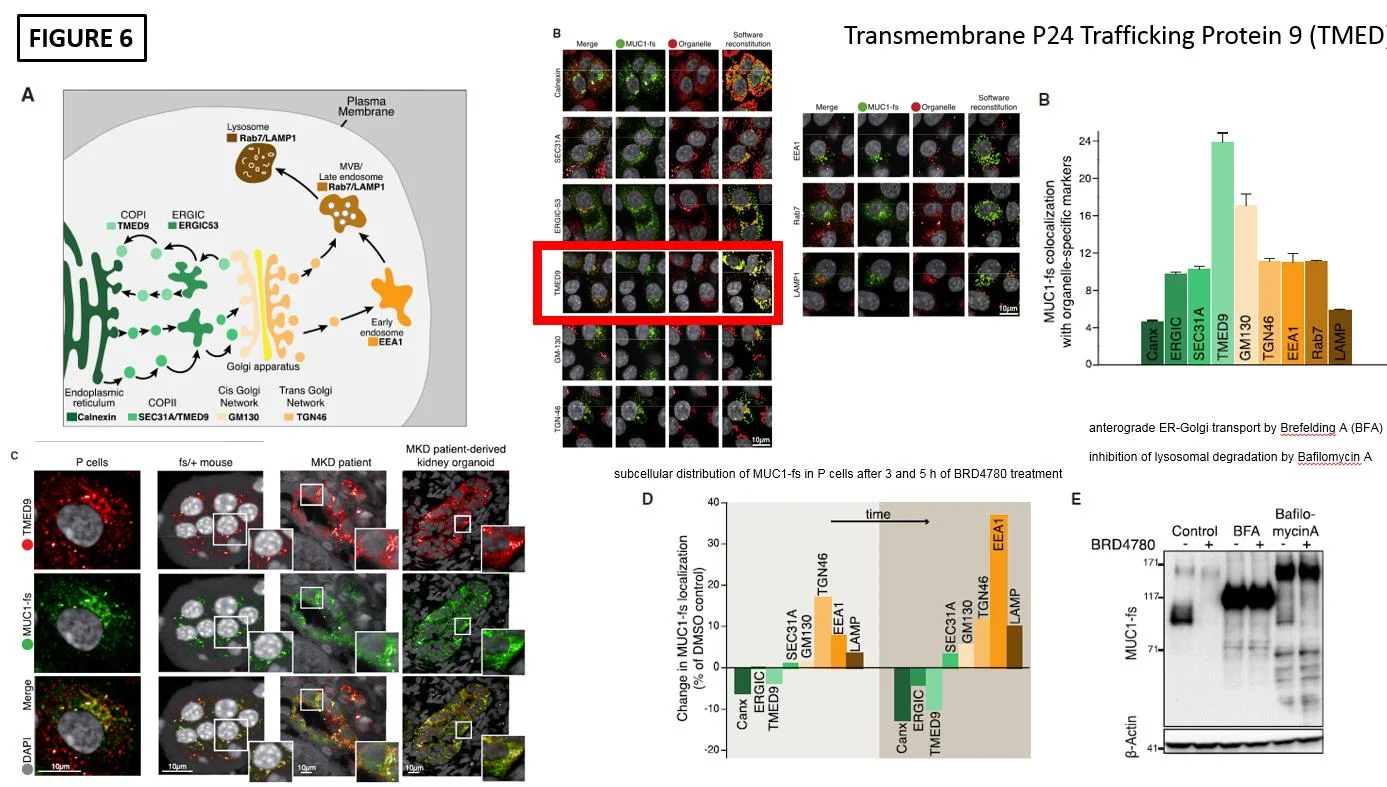
The final piece was to show how exactly BRD4780 was diminishing MUC1-fs. To do this colocalization immunofluorescence was performed. This showed that the highest degree of colocalization of MUC1-fs occured with the cargo protein transmembrane P24 trafficking protein 9 (TMED9) as seen in Figure 6B. After treatment with BRD4780 the colocalization shifted to lysosomal locations for MUC1-fs as seen in Figure 6D. Lastly, Cellular Thermal Shift Assay (CETSA) demonstrated that BRD4780 interacts with TMED9 as seen by a shift upwards, suggesting a potential interaction.
Discussion
The molecular mechanism for certain inherited diseases, caused by accumulation of misfolded proteins in cells, has remained unknown, hence the current lack of therapies. The investigators in this paper have elucidated a potential molecular mechanism on how these diseases are caused. In particular they identified that MUC1-fs accumulates in the cytoplasm of cells. They also recapitulated the human disease in a mouse model. Interestingly the mouse model is worse in female mice. There is no discussion on why this might be. They also demonstrated that the ATF6 pathway of the UPR is upregulated and might serve as a protective mechanism that if overwhelmed leads to cell death. Thus, potentially explains the relatively late (and variable) onset of kidney disease in MKD and the need for a second hit. They identified BRD4780 and demonstrated a potential mechanism of action for the removal of the mutant protein (MUC1-fs), through an interactive with TMED9. The exact interaction of BRD4780 with TMED9 is still unclear and further studies are needed. Although no overt toxicities were seen when mice were treated with BRD4780, TMED9 is expressed in a fairly ubiquitous manner in a variety of cells. Thus, if this interaction with BRD4780 affects other protein interactions and results in toxicity will be important. This discovery has the potential to shed light upon the understanding and development of new drugs for MKD and other toxic proteinopathies. However, several hurdles still exist. For instance, it will be important to see if pathology is reversed with BRD4780 in animal models (and not just clearance of MUC1-fs) and eventually in humans with MKD. Nonetheless, these are impressive studies.
Summary prepared by Vanessa Lerma,
NSMC Intern, Class of 2019