

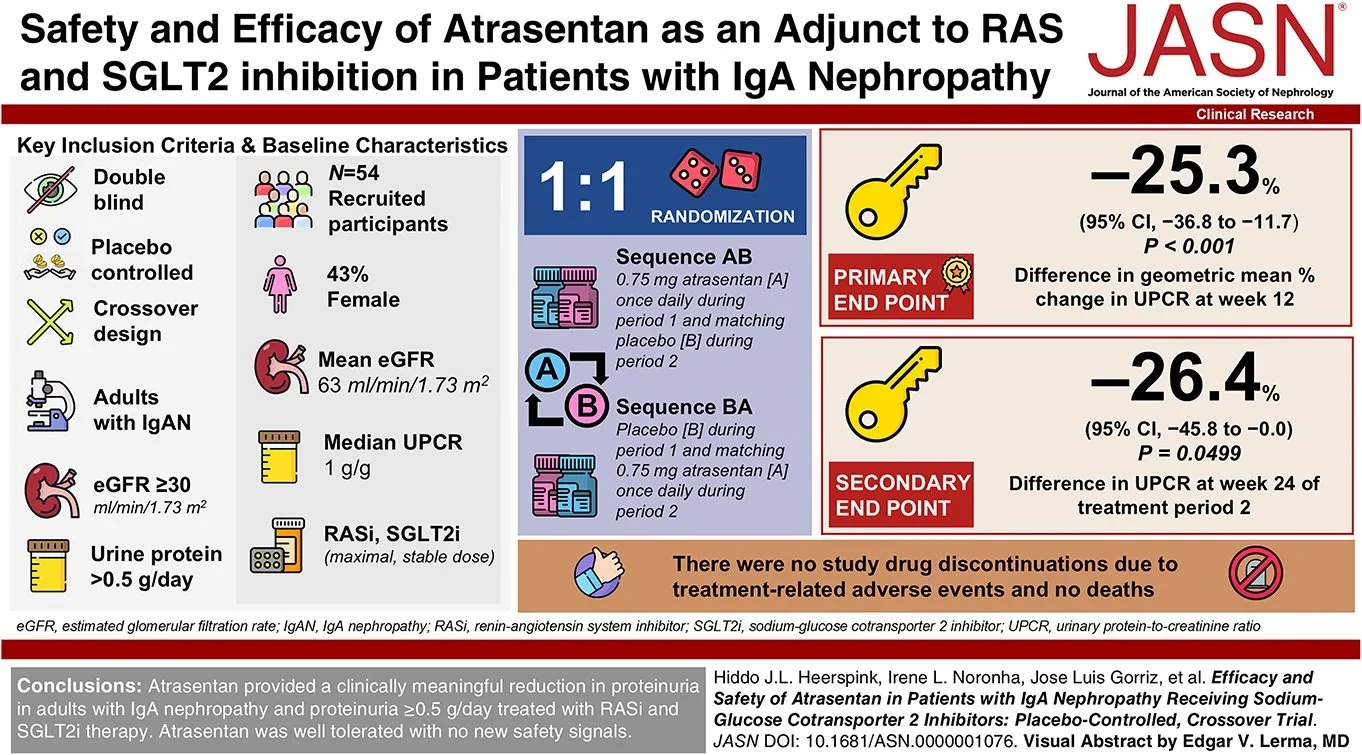
#NephJC Chat
Tuesday, February 11th 2025, 9 pm Eastern on BlueSky
Hypertension. 2025 Feb;82(2):306-318. doi: 10.1161/HYPERTENSIONAHA.124.24153. Epub 2024 Dec 11.
Characterizing the Origins of Primary Aldosteronism
Jenifer M Brown , Brooke Honzel, Laura C Tsai, Julia Milks, Yvonne M Neibuhr, Andrew J Newman, Michael Cherney, David G Stouffer, Richard J Auchus, Anand Vaidya
PMID: 39660429
PMCID: PMC11735322 (available on 2026-02-01)



Introduction
“Idiopathic Hypertension” might bring to mind microvascular gremlins, beyond mere mortal comprehension, suddenly wreaking havoc on the cardiovascular system. However, what if a large number of idiopathic/familial hypertension cases were not secondary to an unelucidated physiologic process but rather the consequence of a subclinical, milder well known disease? Primary aldosteronism (PA), the production of aldosterone independent of renin and angiotensin II (AG II), is a known cause of hypertension that is thought to affect approximately 5-10% of hypertensive patients. In fact, recent studies consistently indicate that PA has been identified in 22% of patients with resistant hypertension and even up to 11% in normotensive patients (Kitamato T, et al. Front Endocrinol 2025). We were all taught in medical school that hyperaldosteronism is associated with the classic triad: elevated blood pressure, hypokalemia and metabolic alkalosis. Many, if not most, clinicians may not even consider a diagnosis of PA without these clinical and/or laboratory findings. In addition, as physicians, we often consider most diseases to be binary (i.e. you have it or you don’t). However, when considering systems that include multiple interactive compounds with complex feedback loops, like the RAAS or glucocorticoid/mineralocorticoid cascades (image 1 and 2), there is the possibility of there being phenotypically mild disease that may not be easily observed by common measures. It is not hard to imagine that there are variations (e.g. isoenzymes) that might lead to slightly worse regulation without complete dysfunction of these systems. Indeed quantifiable processes, like blood pressure and serum metabolic panels, have a range of “normal” values that may not be sensitive enough to trigger concern. This fact may obscure mild alterations that may ultimately have clinical significance due to the accumulation of inflammation and damage over time.
The current study examines the possibility that PA is significantly underrecognized due to mild clinical presentations, and may have greater clinical significance than previously considered in normotensive patients. Recognizing that PA involves a continuum of severity, defined by quantifiable metabolites of aldosterone, may allow for earlier recognition of at risk patients. Early treatment of patients susceptible to PA-related hypertension, vascular stiffness and cardiovascular disease may ultimately improve outcomes.

Image 1. The Renin-Angiotensin-Aldosterone System (RAAS)

Image 2. Adrenal Mineralocorticoid-Glucocorticoid-Androgen pathway
Methods
The authors identified normotensive patients with risk factors for aldosterone dysregulation and future hypertension, but without known primary aldosteronism. Risk factors known to be associated with hyperaldosteronism include increased BMI and patients with elements of the cardiovascular-kidney-metabolic (CKM) disorder.
Patients were recruited from Boston, MA, between July 2018 to October 2022.
Adult patients aged 35-70 of both sexes were eligible for the study. Patients had to be on no treatments for hypertension and have an average BP of 120-135/75-85 mmHg. In addition, patients had to have risk factors for PA including: BMI ≥ 25 kg/m2, or family history of hypertension before age 60 in a parent or sibling plus a personal history of diabetes (A1C <9%, 7<5 mmol/mol) or BP 115-135/70-85. Exclusion criteria included known cardiovascular or cerebrovascular disease, active cancer, eGFR < 60 ml/min, daily opioid/NSAID/glucocorticoid use, or BMI >45 kg/m2.
Study Procedure
To evaluate the degree of renin-independent aldosterone production, all patients underwent an oral sodium suppression test (OSST). Patients consumed at least 200 mmol of sodium and 50-75 mmol of potassium daily for 4-7 days. A 24-hour urine was collected on the last day of the study diet, with measurement of the urinary aldosterone excretion rate (AER), to quantify the severity of the PA phenotype. Blood and urine samples were tested for plasma renin (ELISA), serum aldosterone/serum 18-hybrid steroids (liquid chromatography tandem mass spectrometry), serum and urine electrolytes, AER and tetrahydroaldosterone (electrospray ionization). OSST was validated by a urine sodium excretion > 150 mEq/day and a low plasma renin of < 1 ng/mL/hr (i.e. renin and angiotensin II dependent aldosterone production).

Enzymatic pathway of 11-hydroxylase (CYP11B1) and aldosterone synthase (CYP11B2). Sojaati K, et al. Kid Int, 2004.
Patients also separately underwent a dexamethasone suppression test (DST) and ACTH-stimulation test. Blood testing following DST and ACTH-stim testing included serum aldosterone, cortisol and 18-hybrid steroids.

Patients also underwent non-invasive 24-hour ambulatory BP monitoring. BP recordings were done every 20-30 minutes. Daytime, nighttime and 24-hour average of systolic, diastolic and mean arterial pressures were calculated.
Statistical Analysis
24-hour aldosterone urinary excretion during OSST was categorized into tertiles. Baseline characteristics between tertile groups were compared. Aldosterone, its metabolites, and 18-hybrid steroids obtained during OSST, DST, and ACTH-stimulation were compared across tertiles. Additional testing including 24-hour urine and 24-hour BP measurements, and urine potassium-to-sodium ratios were compared by linear mixed models across tertiles of AER. A heatmap of the spectrum of PA across renin and angiotensin II mediated aldosterone production, ACTH-mediated aldosterone production and markers of mineralocorticoid receptor activation was created.

Figure 4. Heat map of normotensive primary aldosterone patients. Brown J, et al. Hypertension, 2025.
Funding
Awards from American heart Association Career development, and NIH National Heart, Lung and Blood institute.
Results
A total of 62 patents completed the study protocols. Patients had a mean age of 48 years, and BMI of 29.5 (overweight to obese). Blood pressure and serum potassium levels were normal.

Table 1. Baseline Characteristics. Brown J, et al. Hypertension, 2025.
OSST and Independent Aldosteronism
OSST resulted in a mean urine sodium excretion of 258+/-92 mEq/24hrs, and a mean plasma renin of 0.39+/-0.26 ng/mL/hr. There was a spectrum of non-suppressible renin and AG II independent aldosterone levels. This was seen similarly for the related molecule of 18-hydroxycorticosterone, as well as urinary tetrahydroaldosterone.

Figure 1. Renin and AG II independent aldosterone production. Brown J, et al. Hypertension, 2025.
The continuum of renin and AG II independent aldosteronism was also further determined by increases in the intermediate metabolites of 18-hybrid steroid production in the urine. This suggested that there was a corresponding continuum of CYP11B2 activity (non-binary). Even though study patients exhibited “normal BP”, there was a modest but measurably higher systolic daytime BP in patients with higher urinary aldosterone (SBP range by urinary aldosterone tertile in Figure 2D: 102-128 mmHg first, 110-127 mmHg second, 104-138 mmHg third tertiles). Although serum potassium levels were normal, 24-hour urinary potassium/sodium ratios were seen to be increased with each increasing tertile of urinary aldosterone.

Figure 2. Spectrum of associated findings by aldosterone tertiles. Brown J, et al. Hypertension, 2025.
There was also a spectrum of ACTH-independent (post-DST) and ACTH-dependent (post ACTH-stimulation) production of aldosterone and its precursors. Not surprisingly, this was also observed in 18-hybrid steroid production, highlighting a spectrum of ACTH-mediated CYP11B2 activity.

Figure 3. Spectrum of ACTH mediated aldosterone production. Brown J, et al. Hypertension, 2025.
Strengths
-Correlates with multiple national cohort studies which have demonstrated renin-independent aldosterone production is associated with a risk for clinical hypertension and subclinical heart disease
-Rigorous physiological phenotyping
Limitations
-Single center
-Small cohort
-Narrow range of blood pressures make it difficult to differentiate phenotypic difference
-Adrenal imaging, histopathology and genetics were not available
Discussion
Some in the nephrology community believe that hypertensive nephrosclerosis is a fallacy and a cop out diagnosis. In patients diagnosed with hypertensive nephrosclerosis nephrologists are challenged to determine the causal roles of underlying genetics (e.g. APOL1), and more broadly elements contributing to hormonal inflammation and oxidative stress. Sclerosis of renal arterioles, attributed to hypertension and aging, is a frequent finding on kidney biopsies. Hypertension is still the presumed cause of ESRD in as many as 30% of patients in some nephrology textbooks. There is, in fact, a paucity of data showing that moderately elevated blood pressures lead to significant declines in eGFR, or that treating hypertension significantly improves CKD progression (Pajewski N, et al. J Am Soc Nephrol 2024). This leads many to consider the chicken-and-the-egg argument: does hypertension cause CKD or does CKD cause hypertension? In the search for disease processes that are actually the foundational cause of hypertension and kidney disease, subclinical (unrecognized) hyperaldosteronism seems like a plausible explanation.
Aldosterone is a hormone that helps regulate blood pressure and fluid balance. In the kidney, aldosterone is integral in the absorption of sodium and water and the excretion of potassium via ENaC and ROMK. Aldosterone also helps maintain the body’s acid/base balance through pendrin in the ꞵ-intercalated cells of the kidney. As part of the RAAS system aldosterone is a terminal hormone, and is critical to maintaining volume expansion in times of excessive volume loss. But, aldosterone also has a dark side…

Beyond fluid and pH regulation, aldosterone also promotes fibrosis by stimulating collagen production. Aldosterone can induce inflammation and oxidative stress in tissue contributing to vascular and cardiovascular disease (Ardenshorst W, et al. Antioxidants 2024). This indicates that aldosterone could be a main player in cardiovascular remodelling and (most importantly) kidney function. This is where the interplay of mild hypertension and kidney disease may actually intersect. There is even a newly discovered membrane receptor, G protein-coupled estrogen receptor (GPER), that binds aldosterone and has been shown to moderate vasoconstriction, endothelial dysfunction, and apoptosis. (Li X, et al. Front Endocrinol 2023). Mineralocorticoids such as aldosterone trigger a profibrotic process that mimics the early phase of wound healing. Depending on the type of cell involved, aldosterone may activate the profibrotic process through classic mineralocorticoid receptors and nonclassic membrane-associated mineralocorticoid receptors throughout the body. Although sodium in the diet has been implicated in aggravating aldosterone-induced renal fibrotic processes, in reality aldosterone alone can initiate matrix production in renal tissue even in the absence of active sodium transport (Brem A, et al. AJKD 2011). Want to hear more about aldosterone and fibrosis? Dr. Josh Waitzman speaks about this very topic in rodent models on the Channel Your Enthusiasm podcast (Chapter 16 part 2, “Voice of God”: time 39:49-36:57). It is these potential hazards that make MRAs and ns-MRAs one of the main pillars of treatment in both heart and kidney diseases.

From: Aldosterone-Induced Fibrosis in the Kidney: Questions and Controversies. AJKD 2011.
The current study brings to light that aldosterone production, independent of renin and AG II, is a continuum with potentially many people below the threshold of the classic phenotypic findings seen in primary aldosteronism. However, careful measurement of blood pressure, potassium excretion and aldosterone metabolites shows that these individuals could be classified from low to high risk for PA induced hypertension based upon urine and serum markers of aldosterone. Could these subclinical alterations have negative effects at the microvascular level that ultimately lead to overt CKM disease decades later? Again, this seems plausible, and thus earlier recognition of at-risk populations may lead to treatments that prevent cardiovascular and kidney diseases. This may also answer why treating hypertension, once it is clinically evident, seems to fail to curb end organ damage, with declining eGFR, that may have been smoldering for decades. Although PA might be the initial trigger for kidney damage in some patients, ultimately other processes related to inflammation and fibrosis may combine to determine poor outcomes.
As we search for answers and strive for precision medicine in patients with chronic kidney disease, it is heartening that familiar pathologic processes might be at the core of disease initiation. Screening and treatment at the start of inflammation, fibrosis and vascular remodeling may ultimately prove to be one of the keys to CKD prevention. Once they are available, evaluation of genetic markers that allow for screening of subtle PA phenotypes, before any clinical evidence is present, may help tailor therapy in at-risk patients.
Conclusion
Primary Aldosterone represents a continuum of disease that may present sub-clinically, without obvious elevated blood pressures or urinary potassium excretion. Unrecognized, milder phenotypes of PA have the potential for increasing the risk for future clinically significant hypertension, cardiovascular and kidney disease.
Summary Prepared by
Brian Rifkin
Hattiesburg Clinic, Hattiesburg MS
Reviewed by Jamie Willows
Header Image created by AI, based on prompts by Evan Zeitler